
WHAT YOU'LL LEARN
How to diagnose papilledema based on optometric exam and ocular manifestations
Understand the differential diagnoses of papilledema
How to treat and manage papilledema
What to expect from patients presenting with symptoms of papilledema
Introduction
Introduction
Papilledema can be a common neuro-ophthalmic emergency that presents to the primary eyecare provider. This presentation is one of the main reasons patients are referred to the emergency room—to rule out sight/life-threatening conditions such as the presence of an intracranial mass lesion. In this course, we will cover the background, treatment, management, and differential diagnoses of papilledema through the lens of a patient case encountered by Dr. Rahim Bhaloo, OD, FAAO, a member of the National Vision Doctor of Optometry network employed by Texas Eye and Vision Associates, PA and Texas Vision Associates, PA.
Background
The background of papilledema
Papilledema is defined as swelling of the optic disc (almost always bilateral) secondary to increased intracranial pressure (ICP).1 Changes in intracranial pressure can be summarized by the Monro-Kellie hypothesis. Since the total volume within the cranium is fixed, any positive volumetric change in one of the cranial components, either blood, cerebrospinal fluid (CSF), or brain tissue, must be compensated for by a negative volumetric change of another component, otherwise increased ICP will result.2 Until recently, stasis of axoplasmic flow was considered to be the main mechanism of visual loss and optic disc swelling in papilledema.1 Increased ICP subsequently increases CSF pressure throughout the central nervous system, including the subarachnoid space surrounding the optic nerves. This upsets the normal gradient between intraocular pressure and retrolaminar pressure, resulting in higher than expected tissue pressure within the nerves.3,4 It was hypothesized that the resultant increased tissue pressure caused swelling of the optic disc as well as interrupted metabolic processes that mediate axoplasmic flow.5
Two of the most recent theories of papilledema pathogenesis are the mechanical theory and the ischemic theory, and focus on the subarachnoid space of the optic nerve. This subarachnoid space consists of multiple chambers which can dilate and cause compression of the optic nerve. This phenomenon has been associated with papilledema and progressive optic nerve atrophy.6,7
The mechanical theory argues that elevated ICP compresses the optic nerve at the globe junction, causing a backlog of axoplasmic flow at the scleral lamina.8,9 This theory is supported by a study discovering that CSF velocity is markedly slower further away from cerebral ventricles. As a result, any reduction in CSF rate should theoretically affect optic nerve subarachnoid space first, since it is anatomically distant from the ventricles and does not allow for continuous CSF flow.8,10
The ischemic theory insists that vascular flow is compromised secondary to dilation of the optic nerve subarachnoid space. The retrolaminar optic nerve is prone to ischemic insult and therefore raised ICP reduces axonal perfusion.9 This theory coincides with the proposal that the relationship between the choroidal circulation and ciliary arterial circle is responsible for papilledema formation. As subarachnoid pressure increases, ciliary circulation is compressed. This reduces blood flow to the optic nerve, interfering with choroidal blood flow.8 The end result is chronic axonal ischemia which affects metabolic axoplasmic flow.9 Additionally, axonal hypoxia and axoplasmic stasis could lead to the local accumulation of toxic metabolites in the CSF.11
Patient Case
Patient demographics/CC/HPI
A 24-year old Hispanic female presented in office for a routine exam to update her glasses Rx. Her last exam had been 2 years prior. She presented with a chief complaint of headaches x 2 months which she attributed to a change in her distance Rx, as she had noticed changes in her vision. The patient stated that the headaches were non-localized, occurred intermittently throughout the day, and poorly controlled with ibuprofen, and she attributed this to her new desk job that required her to be on a computer 8 hours per day.
Ocular/Medical history
The patient’s ocular history was unremarkable with the exception of low myopia. Her medical history was positive for seasonal allergies, managed with over the counter antihistamines, and she stated that she had no known drug allergies. Patient’s family ocular history was positive for primary open angle glaucoma, and family medical history was positive for hypothyroidism and systemic hypertension. The patient denied alcohol and substance use and denied being sexually active. She stated that besides school her main interest was physical fitness, and she worked part-time as a personal trainer.
Ocular exam
Entering VAs: 20/25 -1 OD, OS, OU with habitual correction of -1.00 DS OD, -1.25 DS OS. Visual acuities did not improve with pinhole. Pupils were equally round and reactive to light and accommodation with no afferent pupillary defect, though it was noted that reaction to light was sluggish OD, OS. Confrontation field testing was full to finger count OD, OS. Color vision testing with HRR plates was 6/7 OD, OS. Extraocular muscle (EOM) testing was unremarkable OD, OS. Cover test with correction showed orthophoria at distance and near. A subjective refraction yielded a best corrected visual acuity of 20/25 OD with -1.00 -0.25 x 010 and 20/25 +1 OS with -1.25 DS. Best correct near visual acuities were 20/20 OD, OS, OU.
Remarkable anterior segment findings included +1 papillae OD, OS and trace diffuse superior punctate keratitis OD, OS. Intraocular pressure by Goldmann applanation tonometry was 19 mmHg OD, 18 mmHg OS at 9:54 AM. One drop tropicamide 1.0% and one drop phenylephrine 2.5% was instilled OU at 9:57 AM to dilate the patient for posterior segment evaluation. While the patient was dilating fundus photography was performed OD, OS.

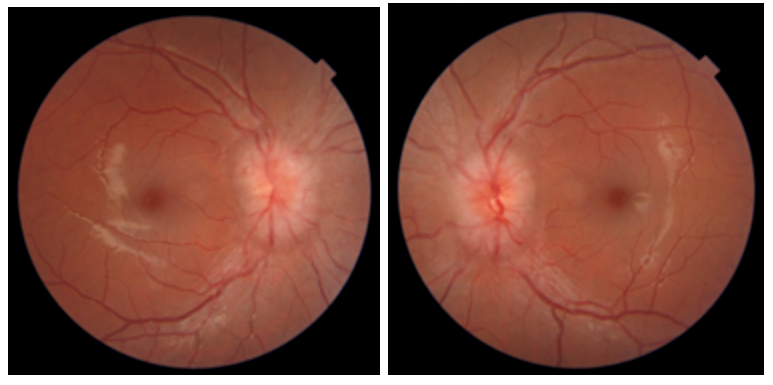
Remarkable posterior segment findings involved the patient’s optic nerve heads and peripapillary areas. Both optic nerves appeared to have 360 degrees of disc elevation with blurred disc margins and blurred obscuration of vessels at the inferior and nasal margins OD, OS. Additionally, the peripapillary area OS exhibited a flame hemorrhage superotemporally. No Paton lines were noted secondary to the nerve edema during evaluation of the posterior segments. It should be noted, however, that this does not definitively exclude their presence, as these peripapillary wrinkles can be obscured by overlying, swollen nerve fibers and are better viewed with optical coherence tomography (OCT).
Upon completion of the exam the patient was immediately referred to the radiology department of the local hospital for magnetic resonance imaging (MRI) with and without T1-weighted contrast of the brain and orbits. The decision to forego a confirmatory OCT was made based on the severity of presumed papilledema, which was quantified as Grade 3 OD, OS on the modified Frisen scale. Radiology reported no abnormalities in the brain of the patient but notated a flattening of the posterior ocular globes. The patient was referred to neurology for further management and underwent a lumbar puncture and extensive bloodwork the same evening. Her opening CSF pressure was 347 mmHg and a week later all blood panels came back unremarkable, confirming increased intracranial pressure of unknown origin. At that point the patient was diagnosed with idiopathic intracranial hypertension.
Treatment and management of patient
The patient began acetazolamide 500 mg bid therapy and was followed closely every 4 weeks for visual field testing and optic nerve OCT. When the patient returned for a complete exam 7 months later, she was free of all visual symptoms with 20/20 distance acuities OD, OS with habitual correction. Anterior and posterior segment evaluations were unremarkable. No appreciable optic nerve edema was viewed with a 90 diopter condensing lens and rim tissue was healthy 360 degrees OD, OS with no evidence of pallor.
Discussion
The Monro-Kellie hypothesis
The Monro-Kellie hypothesis explains the delicate balance of ICP between brain tissue, blood, and CSF volume, and large changes in any of these can cause increased ICP and resultant papilledema. Space-occupying lesions can fall into one of two categories: mass lesions, which can be considered part of the brain tissue component of the Monro-Kelli hypothesis, or cerebral hemorrhages, which are blood components. Mass lesions can be further subdivided into non-infectious lesions, such as tumors, and infectious lesions, such as abscesses.12 Studies dating back decades have postulated that upwards of 80% of patients with cerebral mass lesions can present with papilledema.13 It has also been theorized that infratentorial lesions that obstruct ventricular outflow are more likely to cause papilledema than supratentorial lesions.1 The most common cerebral hemorrhages to cause papilledema are subarachnoid hemorrhages and subdural hematomas.1 Approximately 20% of patients with subarachnoid hemorrhages present with papilledema, and it is thought to be a result of CSF outflow or absorption blockage.1,14 Cerebral trauma has been linked to subsequent rise in ICP, but the correlation of papilledema with trauma-related ICP increase is weak at best. One study found that only 3.5% of patients with ICP elevation secondary to cranial trauma had presented with papilledema.15 Idiopathic intracranial hypertension (IIH) is also a prominent cause of papilledema.16
Common causes of elevated ICP and papilledema
- Cerebral Trauma
- Space occupying lesion – intracranial mass, cerebral abscess, intracranial hemorrhage
- Respiratory failure
- High altitude sickness
- Chiari malformation
- Sarcoidosis
- Leukemia
- Infectious processes – tuberculosis, syphilis, actinomycosis
- Autoimmune – sarcoidosis, Vogt-Koyanagi-Harada (VKH)
- Idiopathic intracranial hypertension (IIH)
- Pharmacological
While intracranial lesions can result in papilledema, very little evidence exists linking intraocular tumors and papilledema. Metastatic lesions most commonly manifest in the uveal tract and subjective visual symptoms are commonly attributed to retinal involvement.17,18 Metastasis to the optic disc can cause optic disc edema,17 but this does not fit the definition of papilledema as the globe itself is not located within the cranial cavity. As such, the ocular globe is not accounted for in the Monro-Kelli hypothesis. While the optic nerve is part of the central nervous system, its meningeal sheath does not extend intraocularly, and therefore intraocular tumors do not interact with CSF. Lesions of the retrobulbar optic nerve, such as a sheath meningioma and oligodendroglioma, have been linked to the development of papilledema since they can impact the flow of CSF as they increase in size.19-21 Since it is assumed that CSF can communicate freely among the different compartments of the central nervous system, the consensus conclusion has been that CSF pressure and concentration is equally affected throughout all compartments.22 This theory is supported by the relative symmetry of optic nerve swelling typically seen in papilledema. Conversely, however, there are many reported cases of asymmetric and even unilateral papilledema.23-27 One study measuring biomarker concentration in CSF discovered vast differences between concentrations in lumbar and optic nerve CSF. This finding suggests that the optic nerve subarachnoid space has the potential to be a separate compartment, which would allow for the development of unilateral papilledema.22
A number of medications have also been shown to be responsible for the development of papilledema. Retinoids such as isotretinoin, used in acne treatment, and all-trans-retinoic acid, which can treat certain types of leukemia, have been postulated to cause increased ICP due to toxicity.28 Excess retinol may be transported into CSF where it can be toxic to the arachnoid granulation resorption mechanism, an important step in CSF reabsorption.29 The tetracycline antibiotics minocycline, tetracycline, and doxycycline have also been associated with increased ICP. While the associated mechanism for ICP increase is not completely understood, it has been theorized that the tetracycline antibiotics inhibit cyclic adenosine monophosphate in the arachnoid granulation resorption complex, thereby decreasing CSF reabsorption.30 Lithium, used in the treatment of bipolar disorder, was first reported to cause papilledema in 1990.29 Multiple mechanisms have been proposed for the development of papilledema secondary to lithium, though no consensus has been reached.31
Synthetic formulations of reproductive hormones such as progestin and estrogen have also been linked to elevated ICP and IIH. A possible correlation has also been found between idiopathic intracranial hypertension (IIH) and polycystic ovarian syndrome (PCOS). Excess estrogen in PCOS has been linked with a prothrombotic state as well as an increase in retinoic acid synthesis, both of which may alter CSF outflow. The estrogen-induced thrombophilia may cause inadequate drainage of CSF due to obstruction of resorption sites.29 Links between synthetic hormone contraceptives and thrombophilia are well established, partly explaining how these agents can cause papilledema32 (as well as the increased preponderance for IIH in younger females). Oral/implantable contraceptives and intrauterine devices have also been linked with IIH and must be addressed accordingly in patients with IIH.
Aldosterone is a mineralcorticoid involved with CSF production. Hyperaldosteronism has also been linked with PCOS and obesity, and may partly explain the correlation with IIH among these patient demographics.
Gleevec, a tyrosine kinase inhibitor used in the treatment of gastrointestinal stromal tumors, has the potential to cause optic nerve edema. One of the primary targets of Gleevec is platelet-derived growth factor (PDGF).33 It has been shown that PDGF is expressed in vascular endothelium and that PDGF inhibition leads to fluid retention in connective tissues and extracellular matrices.34,35 Receptors for PDGF are also located in retinal ganglion cells of the optic nerve.36 One study found that Gleevec-induced inhibition of PDGF could potentiate apoptosis of ganglion cells, and so it has been presumed that this cell apoptosis could be the underlying event causing edema.33,36
Chiari malformation is a group of abnormalities involving cerebellar displacement and spinal column involvement, believed to result from failure of normal dorsal induction in utero.37 Though not as common as its other ocular signs and symptoms, patients with Chiari malformation may present with papilledema.38 It has been postulated that increased ICP and papilledema in Chiari patients are the outcome of obstructed CSF flow inferiorly through the foramen magnum, a direct result of downward displacement of the cerebellar tonsils.39
Papilledema has been found in some individuals suffering from high altitude sickness, or acute mountain sickness (AMS).40 Studies on human and animal subjects have revealed a potential rise in ICP when subjected to hypoxia, and one study found that 59% of climbers evaluated at an elevation of 6,865 meters had some degree of optic disc swelling.41 It has been theorized that compromised venous drainage in hypoxic conditions may account for a hydrostatic pressure change across capillary beds and result in increased ICP, leading to papilledema.40,42,43
Papilledema associated with respiratory failure was first described in 1933.44 Though recent research lacks in this association, the venous theory explaining increased ICP in AMS could explain this mechanism of papilledema (as respiratory failure creates hypoxic conditions). This theory is supported by the correlation of papilledema with other hypoxia-related disorders such as sleep apnea and Pickwickian syndrome.45,46
Infiltrative etiologies like sarcoidosis have been shown to cause papilledema. Sarcoidosis is thought to result from interactions between genetic factors and environmental antigens, and this can lead to the formation of noncaseating granulomas that cause tissue damage in active phases.47,48 Up to 15% of patients with neurosarcoidosis may present with papilledema.49 This can occur as a result of larger intracranial granulomas mimicking tumors or granulomatous meningoencephalitis raising ICP, or by obstructive hydrocephalus arising from granulomas blocking ventricular CSF flow.50
Differential Diagnoses
Idiopathic intracranial hypertension
The first documented case of papilledema from IIH occurred in the late 1800s. IIH is characterized by increased ICP of unknown etiology in the absence of evidence of any intracranial pathology.51 It is a diagnosis of exclusion and has defined diagnostic criteria, also known as the modified Dandy criteria.1,52 The modified Dandy is comprised of the following findings: signs or symptoms explained by increased ICP such as headache, pulsatile tinnitus, papilledema, or a non-localizing CN VI palsy; neuroimaging of the brain and orbits negative for a space-occupying lesion, hemorrhage, hydrocephalus, or venous sinus thrombosis; elevated opening CSF pressure by LP in the left lateral decubitus position with normal CSF composition; and no alternative underlying findings.53,54
The most common ocular manifestation of IIH is papilledema.55 Papilledema pathogenesis in IIH is not completely understood. Though the Monro-Kellie hypothesis provides a guideline for the development of increased ICP, five main hypotheses exist which try to explain its mechanism in IIH:
- A reduction in CSF reabsorption at arachnoid granulations is known to cause elevated ICP by either increased CSF outflow resistance or pressure gradient reduction between subarachnoid space and the superior sagittal sinus.53 This is the same mechanism responsible for increased ICP secondary to hemorrhage. In IIH patients, reduced CSF reabsorption has been demonstrated using radioactive markers to track CSF circulation.56 One contradiction to this theory is that arachnoid granulations do not exist prenatally; therefore, a different pathway for CSF absorption is critical during fetal development.57,58 This theory was supported by an experiment that demonstrated a link between lymph and CSF in primate nasal mucosa.59 The findings of this experiment led to the belief that CSF absorption is driven by the lymphatic system at low pressure and arachnoid granulations at high pressure.60 Additionally, consideration of Starling’s forces and its effect on CSF regulation has led to the belief that cerebral capillaries play a major role in the reabsorption of CSF.61
- Advances in venographic imaging have shown that most IIH patients have cerebral venous sinus system abnormalities, including stenosis of the transverse sinuses.62 Though no consensus yet exists, it seems as though this stenosis is a consequence of increased ICP and not a congenital risk factor.63
- It is thought that excessive secretion of CSF by the choroid plexus (or abnormal CSF reabsorption through the subarachnoid space) leads to elevated ICP and IIH. Approximately 600mL of CSF is produced daily and gradually decreases over time. Initial suggestions regarding CSF production labeled the choroid plexus as the main contributor, with two-thirds of CSF secretion believed to have been located at the plexus and regulated by ion transporters on plexus epithelial cells.64 It was therefore hypothesized that increased CSF production could be linked to increased plexus size or increased activity of ion transporters, and that this could be an underlying root cause of increased ICP. However, patients diagnosed with IIH have not typically shown signs of plexus hypertrophy, and CSF overproduction has only been demonstrated in disorders that cause hydrocephalus.53 Since the presence of hydrocephalus contradicts an IIH diagnosis, this is no longer a reasonable theory. Furthermore, after it was reported that capillary surface area in the brain was about 5,000 times larger than that of the choroid plexus, recent studies have theorized that CSF is primarily produced in the capillaries of the cerebral parenchyma.61,65,66 This theory demonstrates that, if CSF volume is regulated within cerebral capillaries by hydrostatic and osmotic pressure differences (Starling’s forces), then elevated ICP will actually decrease CSF production.61,67 This contradicts the previous hypothesis of the choroid plexus as a main CSF production site and further weakens the theory correlating plexus hypertrophy with increased ICP. While the literature is sparse with definitive evidence on excessive CSF production in IIH, increased resistance to CSF reabsorption has been established in cases of IIH. The decrease in CSF production with age may partly explain why IIH primarily occurs in younger demographics.
- The positive correlation between obesity and IIH development is well documented.55,68-71 Unfortunately, no pathological mechanism connecting the two has been substantiated.53 It has been hypothesized that elevated intra-abdominal pressure in obesity can lead to increased ICP via the venous vessels in the spine. However this doesn’t entirely explain the marked female predominance in IIH.
- The role of gender is currently being studied with regards to IIH. IIH typically affects overweight women of child-bearing age.55 One long-term study estimated an overall IIH incidence rate of 2:100,000 in the general population.72 Comparatively, recent studies in the United Kingdom and United States among obese women found IIH incidence rates of 11.9:100,000 and 323:100,000, respectively.73,74
IIH Symptoms
Up to 96% of patients with IIH will have some changes to their vision. This primarily occurs by increased intracranial pressure (ICP) significantly compromising axoplasmic flow along the visual axis, leading to variable changes in vision. Since the optic nerve is surrounded by the meninges and cerebral spinal fluid (CSF), it is directly affected by elevated ICP. In IIH, the relationship between intraocular pressure and retrolaminar pressure is altered. Common ocular manifestations of IIH include transient visual obscuration, blurry vision or vision loss, and diplopia.53,55 It is important to remember, however, that none of these symptoms are pathognomonic for IIH.75 Transient visual obscuration lasting seconds can be precipitated by changes in posture. It is thought that episodes of transient ischemia to the optic nerve head, combined with reduced optic disc perfusion pressure, can occur during sudden fluctuations in ICP, causing visual obscurations.16,53 Blurred vision and visual loss are a direct result of acute and chronic papilledema. In the acute stage, a swollen optic nerve head most commonly creates an enlarged blind spot, and this is often the only visual field change.76 In chronic or severe papilledema nerve fiber damage can occur at the level of the optic disc.77 These are the same nerve fiber bundles that are affected in glaucoma, and as such nasal steps and arcuate defects are not uncommon to see during perimetry testing.16,78 The papillomacular bundle, and therefore central visual acuity, is typically spared until late in the disease process.79 Late- and end-stage papilledema can mimic resolution as progressive and consistent axoplasmic flow stasis causes atrophy to the nerve; an atrophic nerve will not swell, even during recurrences of increased ICP.16,78
Foster Kennedy vs. pseudo-Foster Kennedy syndrome
Foster Kennedy Syndrome exists when an intracranial mass causes compressive, progressive atrophy to ipsilateral optic nerve, and contralateral papilledema.80 Contralateral papilledema in Foster Kennedy Syndrome is explained by the Monro-Kellie hypothesis. Though ipsilateral papilledema may exist in early stages, a rapid rise in intracranial pressure which then remains elevated can cause post-papilledema optic atrophy within a matter of days, before signs of chronic papilledema set in.81 The absence of an intracranial mass with neuro-imaging can eliminate Foster Kennedy Syndrome as a potential diagnosis of papilledema.
Pseudo-Foster Kennedy Syndrome presents with the same ocular findings as its name-sake, however, in absentia of an intracranial mass. Though rare, the most common associated pathologies presenting as Pseudo-Foster Kennedy Syndrome have been sequential optic neuritis and sequential ischemic optic neuropathy, which cause the progressive unilateral atrophy mimicking a compressive lesion.82-84 Pseudo-Foster Kennedy Syndrome has also been described in the presence of unilateral optic nerve hypoplasia.85
Optic nerve head (ONH) drusen
ONH drusen are calcified hyaline bodies located within the optic nerve.86 These optic nerve anomalies are benign and usually bilateral. Buried ONH drusen may give an appearance similar to optic nerve edema and can be diagnosed with OCT, fundus autofluorescence (FAF), B-scan ultrasonography, or fluorescein angiography (FA).87 Most patients with ONH drusen are asymptomatic but the finding can be associated with acute visual loss.86 ONH drusen have been associated with smaller, crowded nerves. Though the pathogenesis of ONH drusen formation is not explicitly known, it has been postulated that these crowded nerves may propagate drusen formation secondary to abnormal vasculature, which allows for transudation of plasma proteins and deposition of extracellular materials, including calcium-rich mitochondria.88,89 Abnormal axonal metabolism related to this abnormal vasculature can cause calcification of the mitochondrial bodies, creating and growing ONH drusen.90 Patients with ONH drusen are at high risk for developing non-arteritic anterior optic neuropathy (NAION).86
Non-arteritic anterior ischemic optic neuropathy (NAION)
NAION is an acute event arising from small vessel infarction of the anterior portion of the optic nerve.91 NAION is typically unilateral and occurs in patients 50 years and older. The largest risk factors for NAION are a small, crowded optic disc with or without drusen, and vascular risk factors such as diabetes or systemic hypertension.92 The International Optic Neuropathy Decompression Trial (IONDT) highlighted that 60% of NAION patients had at least one vascular risk factor at the time of onset, but there has been no histological confirmation that these vascular risk factors play a role in the pathogenesis of NAION.93,94 In patients under the age of 50, hypercholesteremia has been found to be a risk factor for the development of NAION, which may be the first sign of elevated serum lipids.95 Compared to patients that suffer from NAION in the absence of ONH drusen, patients with an occurrence of NAION in the presence of ONH drusen are typically younger, have had transient visual symptoms prior to the ischemic event, and have more favorable visual prognoses.96 Causes of bilateral NAION include acute hypovolemia, hyperhomocysteinemia, sildenafil use, and sequela of non-ocular surgery.97-100
Vogt-Koyanagi-Harada disease
VKH is a not-well understood autoimmune disorder that can affect multiple systems.101 Though bilateral panuveitis is the most common ocular finding in patients with VKH, bilateral NAION is a common finding.102-105 While the pathogenesis of VKH is not known with certainty, studies have shown that individuals with the HLA-DR4 gene are predisposed to a specific adaptive immune response. This response is expressed by melanocytes in multiple organs, resulting in an autoimmune disease affecting multiple systems.106
Infectious process
Overview
Bilateral ONH edema has been reported in infectious cases of Lyme disease and HIV.107 To test for the presence of Borrelia burgdorferi, the causative agent of Lyme disease, Lyme antibody blood tests measuring the levels of immunoglobulins G and M, as well as a Lyme Western Blot should be ordered.108 To test for the presence of HIV blood antibody testing is also performed, in this case for HIV-1/HIV-2 enzyme by means of immunoassay.109
Papilledema has been found to be the most common ocular abnormality in tuberculosis-associated meningitis.110 Diagnosis for tuberculosis has historically been done with a combination of a skin test, sputum analysis, and often chest x-ray, but more recently nucleic acid amplification testing has drastically reduced the amount of time needed to confirm a diagnosis.111,112
Though progression to neurosyphilis is uncommon, findings of papilledema are seen in approximately 13% of patients.113 Syphilis can be diagnosed with a combination of venereal disease research laboratory (VRDL) and rapid plasma regain (RPR) testing.114
Papilledema has been seen in intracranial actinomycosis, which should be diagnosed by histological evaluation and bacterial culture of affected tissue.115,116 As well, although rare, Whipple’s disease has been linked with bilateral optic disc swelling.117 Diagnosis is made by histological evaluation of small intestine biopsy and polymerase chain reaction.118
Leukemia
Leukemia is a malignant neoplasm of the bone marrow resulting in excessive production of leukocytes, which in turn affect the body’s organs.119 It is one of the most common neoplasms and can be acute or chronic.120 The acute forms of the malignancy are acute myelogenous leukemia (AML) and acute lymphocytic leukemia (ALL), and they can be distinguished by histological evaluation of bone marrow biopsy combined with immunophenotyping by flow cytometry of the bone marrow samples. Posterior ocular signs and symptoms, primarily hemorrhages, Roth spots, and papilledema, occur in up to 50% of patients with ALL and AML, and can sometimes precede systemic features of the malignancy.119,121 Multiple case studies have reported on the finding of bilateral papilledema in AML patients.122,123 It has been postulated that papilledema in acute leukemia may be the result of either leukemic infiltration of the eye and optic nerve, or secondary due to intracranial hypertension produced by therapeutic and immunosuppressive agents.124,125 One study of pediatric leukemia showed that 75% of patients that had papilledema were currently undergoing treatment with pharmaceutical agents known to cause papilledema.125
Hormone imbalance
Thyroid disease, Addison’s disease, and hypoparathyroidism have also been shown to cause bilateral ONH edema.107 Blood tests assessing the hormone levels regulated by the thyroid and parathyroid can shed light on whether or not these organs are functioning properly. Addison’s disease, or adrenal insufficiency, can be diagnosed by checking for low sodium and high potassium levels in urine in conjunction with serum cortisol levels below 100 nmol/l.126 ONH edema is not likely a presenting ocular sign of Addison’s disease, with the most common symptoms being malaise, loss of appetite, salt craving and nausea.127
Ocular manifestations
Overview
A patient suffering from true papilledema can present with many subjective visual symptoms. While there is no guarantee that a patient will complain about all (or any) of the following, it is possible for the papilledema patient to also be suffering from blurred vision, transient obscuration, pulsatile tinnitus, diplopia, and visual field defects.1,51 Cranial nerve (CN) anatomy plays a role in the development and progression of these symptoms. Four of the twelve cranial nerves in particular are vulnerable due to increased ICP: CNs II, III, IV, and VI.
CN II, the optic nerve, is the only cranial nerve that is an extension of the central nervous system.128 It is, therefore, contained within a meningeal sheath and surrounded by CSF making it sensitive to increases in ICP. Studies have shown that in addition to papilledema, increased ICP can cause damage to the lamina cribrosa as well as nerve fiber bundles at the layer of the optic disc.12,76,129
CN III, the oculomotor nerve, is vulnerable to increased ICP secondary to cerebral herniation. Neurons of CN III originate in the midbrain and exit through the interpeduncular cistern, between the posterior cerebral artery (PCA) and superior cerebellar artery (SCA).128 As CN III exits the midbrain, it is susceptible to stretching by potential herniation of the medial temporal lobe through the tentorial notch in the presence of increased ICP.12 Compression of CN III would manifest either as ptosis, horizontal diplopia, or pupil irregularities. CN IV, the trochlear nerve, is also susceptible to tentorial herniation as its path from the pons to the superior oblique muscle includes the free margin of the tentorium.12 Compression of CN IV would manifest as vertical diplopia.
CN VI, the abducens nerve, originates from the dorsal pons inferior to the fourth ventricle.130 Between its exit at the brainstem and entrance to the central venous portion of the cavernous sinus, CN VI transverses through subarachnoid space, leaving it particularly vulnerable to increased ICP as it crosses Dorello’s canal.131-133 After CN II, it is the most likely nerve to be affected by an increase in ICP, with multiple studies calculating an approximate CN VI palsy incidence rate of 12% in patients with IIH.133,134 Stretching of CN VI would manifest as horizontal diplopia. CN VI is also vulnerable due to its close proximity to the clivus.135 The clivus forms the anterior boundary of the posterior cranial fossa and so disorders causing herniation through this space, such as Chiari malformation, can affect CN VI.136 Bilateral involvement of CN VI will most likely indicate a disturbance in this region.135
Work-up
Overview
The standard work-up for papilledema includes a combination of neuro-imaging, lumbar puncture (LP), lab work, and more recently, ocular ultrasound. Prior to these tests, a number of in-office tests can help begin to differentiate a diagnosis. If papilledema is suspected, a thorough medical history and questionnaire of common papilledema-associated symptoms could provide diagnostic clues. Visual acuity and color vision are unlikely to be affected early in the disease process; however, visual field defects manifest early, with an enlarged blind spot as the most common defect observed.137,138 Pupillary light reflexes may be sluggish in patients with papilledema, though an afferent pupillary defect is more likely to be associated with optic neuropathy.137 An optical coherence tomography (OCT) can be performed in-office and can confirm a diagnosis if the retinal pigmented epithelium is deflected anteriorly towards the vitreous humor.139
The first referred test to be performed is neuro-imaging. If papilledema is caused by IIH, common findings seen by magnetic resonance imaging (MRI) include tortuous optic nerves, an empty sella turcica, and flattening of the posterior sclera.140 While the preferred standard of care is MRI, patients with acute presentations may be scanned with computerized tomography (CT) of the brain and CT venography to quickly rule out a sinus thrombosis or space occupying lesion.141,142 Ruling out space occupying lesions as a cause for increased ICP is vitally important before performing LP, as performing LP in this setting can precipitate cerebral herniation, in turn leading to neurological deterioration that can be fatal.143-145
If neuro-imaging does not uncover positive findings for the cause of papilledema, LP should be performed. An abnormally high finding of CSF opening pressure can be expected in patients whose papilledema is a result of IIH. Recent studies have shown that an opening CSF measurement of greater than 250 mmH₂O can confirm a diagnosis of IIH in adult patients, but that this number should be adjusted downwards to approximately 180 mmH₂O in pediatric populations.146-148 Marginally high opening pressure measurements may be found in obese patients in the absence of IIH, but a measurement above 280 mmH₂O should always be treated as abnormal.55 It should be noted, however, that factors including posture and anesthesia effect can affect opening pressure measurements.149 Additionally, one recent study found that when compared to intracranial pressure monitoring, LP measurements tended to overestimate actual readings.150
Recently the use of ocular B-scan ultrasonography has begun to become more widespread as a useful diagnostic tool to differentiate true papilledema from pseudopapilledema. Pseudopapilledema mimics papilledema in that the optic nerve head appears to be elevated, but no peripapillary edema exists. This finding can appear in patients with conditions such as hyperopia, optic neuritis, and optic nerve head drusen.151 B-scan ultrasonography is particularly useful in confirming the presence of buried drusen within the nerve head. This is because the ultrasonography has a strong ability to demonstrate calcium deposits, a primary component of drusen, with a low false positive rate.152 If a patient is sent directly for neuro-imaging it’s possible small, buried drusen could be missed, as MRI does not offer much diagnostic value for nerve drusen, while CT can also miss smaller nerve drusen due to window thickness limitations.153,154
Not only is ocular B-scan ultrasonography useful at finding buried optic nerve drusen, but it is also an efficient technique for measuring optic nerve sheath diameter non-invasively and in-office. Multiple studies agree that optic nerve sheath diameter of greater than 5 mm posterior to the optic globe is abnormal.155-157 Larger measurements found on ocular B-scan ultrasonography can aid in the determination of true papilledema, and many studies have correlated enlarged optic nerve sheath diameters with a diagnosis of IIH.158-161
Laboratory testing of blood and serum can be beneficial in helping obtain an explanatory diagnosis for papilledema if the underlying cause is an infectious agent, nutritional deficiency, or hormonal disorder.
Treatment and management
Overview
The treatment and management of papilledema can be approached both surgically and non-surgically.
Surgical
Transverse venous sinus stenting has been shown to be an effective method of lowering ICP and thereby aiding in the resolution of papilledema. Though debated, it seems venous sinus stenosis is a consequence of increased ICP and not a congenital or pre-symptomatic finding.62 A recent meta-analysis showed that for patients undergoing the stenting procedure 93.7% showed an improvement of papilledema, with symptom recurrence rate requiring additional stenting in 9.8% of patients.162
CSF diversion by lumboperitoneal shunting (LPS) has shown to be effective in symptom reduction for patients with IIH since the 1970s. A catheter is inserted through the lumbar spine into the subarachnoid space, where it collects CSF and funnels it to the peritoneal cavity for absorption.53 Though the procedure has been shown to be effective in resolution of symptoms in patients with visual complaints, it has been reported that 51% of patients have required at least one shunt revision.163
Optic nerve sheath fenestration is effective in relieving the consequences of raised CSF pressure by making incisions to the meninges that enclose the optic nerves.53 One recent meta-analysis calculated that, on average, patients undergoing this procedure had an 80% reduction in papilledema, though repeat surgery was required in 14.8% of patients.164 Surprisingly, evidence exists that sheath fenestration may only need to be performed on one nerve to produce bilateral results.165 However, sheath fenestration is believed to be a temporary measure to preserve optic nerve health and function without being a cure of IIH.16
In patients with IIH a small body of evidence exists showing the efficacy of gastric bypass surgery. Since a relationship exists between obesity and IIH, it makes sense that surgery to treat the obesity may aid in the resolution of IIH.166 In one review article, it was found that 97% of obese IIH patients that underwent gastric bypass surgery saw complete resolution of papilledema.167
In the case of papilledema secondary to a space occupying lesion, a procedure to address the causative agent will result in, in the absence of complications, the lowering of ICP as dictated by the Monro-Kellie hypothesis.
Non-surgical
The first documented study of diet as treatment for IIH and papilledema occurred in 1974.168 Further studies confirmed the correlation between weight loss and papilledema improvement,169,170 and it has been quantified that weight reduction of 6% has been associated with improvement in IIH-related papilledema.28,169 However, since weight loss is not immediate, pharmacologic treatment is still indicated at initial presentation.55
Acetazolamide is a carbonic anhydrase inhibitor that reduces CSF secretion by impeding activity at the choroid plexus.53 It has been researched thoroughly as front-line therapy in patients with IIH and is typically dosed 500mg BID. Unfortunately, some patients may require long term treatment with acetazolamide over months to years. Patients with a history of renal disease may require a smaller dose. The Idiopathic Intracranial Hypertension Treatment Trial (IIHTT) studied the effectiveness of acetazolamide therapy combined with weight loss for improvement of IIH symptoms. The IIHTT found that acetazolamide therapy in IIH patients is associated with improvement of papilledema, but did acknowledge that the subjective nature of the modified Frisen scale for papilledema grading remains a limitation in the quantification of papilledema improvement.171 Furosemide, a weak carbonic anhydrase inhibitor, has also been shown to have some effectiveness in the treatment of IIH and its symptoms when acetazolamide has failed or is contraindicated.16,53
Corticosteroids have been used in the past to treat papilledema, and while dated studies have shown their effectiveness in papilledema resolution, caution is recommended with long term therapy.172-174 Recurrence of papilledema and rebound ICP elevation are possible with dose tapering, and long-term use of corticosteroids has been associated with weight gain and salt retention, which have both been anecdotally linked to worsening symptoms of IIH.16,55
Topiramate, a voltage-gated channel inhibitor, has been found to prominently aid with weight loss in IIH patients, which can have a positive effect on papilledema resolution.53 Its neurocognitive side effects have limited its use, however, and the agent is not recommended as a first-line therapy or in patients with a history of depression.16,53
Conclusion
Papilledema is specific optic nerve edema related to the disruption of a delicate balance within the cranial cavity. Papilledema can present independently or with numerous subjective ocular symptoms, and therefore is often uncovered during a comprehensive exam with an optometrist. A combination of in-office imaging, neuro-imaging, and invasive testing can diagnose the underlying condition causing papilledema, at which point timely therapy can be initiated. Whether surgical or pharmacological, precise and effective treatment can help stabilize intracranial pressure and preserve the integrity of the optic nerve while also correcting a neurological abnormality.
References
- Rigi, M, Almarzouqi, SJ, Morgan, ML, et al. Papilledema: epidemiology, etiology, and clinical management. Eye Brain 2015; 7: 47-57.
- Wilson, MH. Monro-Kellie 2.0: the dynamic vascular and venous pathophysiological components of intracranial pressure. J Cereb Blood Flow Metab 2016; 36(8): 1338-50.
- Hayreh, SS. Pathogenesis of optic disc oedema in raised intracranial pressure. Trans Ophthalmol Soc UK 1976; 96(3): 404-07.
- Hayreh, SS. Optic disc edema in raised intracranial pressure. V. Pathogenesis. Arch Ophthalmol 1977; 95(9): 1553-65.
- Hayreh, SS, March, W, Anderson, DR. Pathogenesis of block of rapid orthograde axonal transport by elevated intraocular pressure. Exp Eye Res 1979; 28(5): 515-23.
- Mashima, Y, Oshitari, K, Imamura, Y, et al. High-resolution magnetic resonance imaging of the intraorbital optic nerve and subarachnoid space in patients with papilledema and optic atrophy. Arch Ophthalmol 1996; 114(10): 1197-203.
- Eliseeva, NM, Serova, NK, Arutiunov, NV. Magnetic resonance imaging of the orbital portion of the optic nerve at different stages of papilledema. Vestn Oftalmol 2005; 121(6): 5-9.
- Passi, N, Degnan, AJ, Levy, LM. MR imaging of papilledema and visual pathways: effects of increased intracranial pressure and the pathophysiologic mechanisms. AJNR Am J Neuroradiol 2013; 34(5): 919-24.
- Trobe, JD. Papilledema: the vexing issues. J Neuroophthalmol 2011; 31(2): 175-86.
- Howden, L, Giddings, D, Power, H, et al. Three-dimensional cerebrospinal fluid flow within the human ventricular system. Comput Methods Biomech Biomed Engin 2008; 11(2): 123-33.
- Killer, HE, Jaggi, GP, Miller, NR. Papilledema revisited: is its pathophysiology really understood? Clin Exp Ophthalmol 2009; 37(5): 444-7.
- Roytowski, D, Figaji, A. Raised intracranial pressure: what is is and how to recognise it. CME 2013; 31(3): 85-90.
- Petrohelos, MA, Henderson, JW. The ocular findings of intracranial tumor; a study of 358 cases. Am J Ophthalmol 1951; 34(10): 1387-94.
- Fahmy, JA. Papilloedema associated with ruptured intracranial aneurysms. Acta Ophthalmol (Copenh) 1972; 50(6): 793-802.
- Selhorst, JB, Gudeman, SK, Butterworth, JF 4th, et al. Papilledema after acute head injury. Neurosurgery 1985; 16(3): 357-63.
- Lee, AG, Wall, M. Papilledema: are we any nearer to a consensus on pathogenesis and treatment? Curr Neurol Neurosci Rep 2012; 12(3): 334-39.
- Konstantinidis, L, Damato, B. Intraocular metastases – a review. Asia Pac J Ophthalmol (Phila) 2017; 6(2): 208-14.
- Konstantinidis, L, Rospond-Kubiak, I, Zeolite, I, et al. Management of patients with uveal metastases at the Liverpool ocular oncology center. Br J Ophthalmol 2014; 98(1): 92-98.
- Roncone, DP. Papilloedema secondary to oligodendroglioma. Clin Exp Optom 2016; 99(6): 507-17.
20. Parker, RT, Ovens, CA, Fraser, CL, et al. Optic nerve sheath meningiomas: prevalence, impact, and management strategies. Eye Brain 2018; 10: 85-99.
21. Chin, HW, Hazel, JJ, Kim, TH, et al. Oligodendrogliomas: a clinical study of cerebral oligodendrogliomas. Cancer 1980; 45: 1458-66.
22. Killer, HE, Jaggi, GP, Flammer, J, et al. The optic nerve: a new window into cerebrospinal fluid composition? Brain 2006; 192(Pt 4): 1027-30.
23. Killer, HE, Laeng, HR, Groscurth, P. Lymphatic capillaries in the meninges of the human optic nerve. J Neuroophthalmol 1999; 19(4): 222-8.
24. Killer, HE, Laeng, HR, Flammer, J, et al. Architecture of arachnoid trabeculae, pillars, and septa in the subarachnoid space of the human optic nerve: anatomy and clinical considerations. Br J Ophthalmol 2003; 87(6): 777-81.
25. Killer, HE, Flammer, J. Unilateral papilledema caused by fronto-temporo-parietal arachnoid cyst. Am J Ophthalmol 2001; 132(4): 589-91.
26. Chang, T, Alibhoy, AT. Unilateral papilledema. Pract Neurol 2017; 17(4): 310-11.
27. Kulkarni, GB, Singh, RJ, Gadad, V, et al. Unilateral papilledema in cerebral venous sinus thrombosis. J Neurosci Rural Pract 2017; 8(Suppl 1): S106-S110.
28. Warner, JEA, Bernstein, PS, Yemelyanov, A, et al. Vitamin A in the cerebrospinal fluid of patients with and without idiopathic intracranial hypertension. Ann Neurol 2002; 52(5): 647-50.
29. Thon, OR, Gittinger, JW Jr. Medication-related pseudotumor cerebri syndrome. Semin Ophthalmol 2017; 32(1): 134-43.
30. Ang, ERG, Zimmerman, JCC, Malkin, E. Pseudotumor cerebri secondary to minocycline therapy. Eur Neurol 1978; 17(1): 48-9.
31. Levine, SH, Puchalski, C. Pseudotumor cerebri associated with lithium therapy in two patients. J Clin Psychiatry 1990; 51(6): 251-3.
32. Tchaikovski, SN, Rosing, J. Mechanisms of estrogen-induced venous thromboembolism. Thromb Res 2010; 126(1): 5-11.
33. DeLuca, C, Shenouda-Awad, N, Haskes, C, et al. Imatinib mesylate (Gleevec) induced unilateral optic disc edema. Optom Vis Sci 2012; 89(10): e16-e22.
34. Lindahl, P, Johansson, BR, Leveen, P, et al. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997; 277(5323): 242-5.
35. Kwon, SI, Lee, DH, Kim, YJ. Optic disc edema as a possible complication of Imatinib mesylate (Gleevec). Jpn J Ophthalmol 2008; 52(4): 331-33.
36. Biswas, SK, Zhao, Y, Sandirasegarane, L. Imatinib induces apoptosis by inhibiting PDGF- but not insulin-induced PI 3-kinase/Akt survival signaling in RGC-5 retinal ganglion cells. Mol Vis 2009; 15: 1599-610.
37. Hadley, DM. The Chiari malformations. J Neurol Neurosurg Psychiatry 2002; 72(Suppl II): ii38-ii40)
38. Choudhari, KA, Cooke, C, Tan, MH, et al. Papilloedema as the sole presenting feature of Chiari I malformation. Br J Neurosurg 2002; 16: 398-400.
39. Mirone, G, Cinalli, G, Spennato, P, et al. Hydrocephalus and spinal cord tumors: a review. Childs Nerv Syst 011; 27: 1741-49.
40. Wilson, MH, Wright, A, Imray, CH. Intracranial pressure at altitude. High Alt Med Biol 2014; 15(2): 123-32.
41. Bosch, MM, Barthelmes, D, Merz TM, et al. High incidence of optic disc swelling at very high altitudes. Arch Ophthalmol 2008; 126(5): 644-50.
42. Wilson, MH, Newman, S, Imray, CH. The cerebral effects of ascent to high altitudes. Lancet Neurol 2009; 8(2): 175-91
43. Wilson, MH, Davagnanam, I, Holland, G, et al. Cerebral venous system and anatomical predisposition to high-altitude headache. Ann Neurol 2013; 73(3): 381-9.
44. Pye, IF, Blandford, RL. Papilloedema associated with respiratory failure. Postgrad Med J 1977; 53(625): 704-9.
45. Reeve, P, Harvey, G, Seaton, D. Papilloedema and respiratory failure. Br Med J (Clin Res Ed) 1985; 291(6491): 331-2.
46. Varelas, PN, Spanaki, MN, Rathi, S, et al. Papilledema unresponsive to therapy in Pickwickian syndrome: another presentation of pseudotumor cerebri? Am J Med 2000; 109(1): 80-1.
47. Matsou, A, Tsaousis, KT. Management of chronic ocular sarcoidosis: challenges and solutions. Clin Ophthalmol 2018; 12: 519-32.
48. Kiszalkiewicz, J, Piotrowski, WJ, Brzezianska-Lasota, E. Selected molecular events in the pathogenesis of sarcoidosis – recent advances. Penumonol Alergol Pol 2015; 83(6): 462-75.
49. Pasadhika, S, Rosenbaum JT. Ocular sarcoidosis. Clin Chest Med 2015; 36(4): 669-83.
50. Katz, JM, Bruno, MK, Winterkorn JM, et al. The pathogenesis and treatment of optic disc swelling in neurosarcoidosis: a unique therapeutic response to infliximab. Arch Neurol 2003; 60(3): 426-30.
51. Kanagalingam, S, Subramanian, PS. Update on idiopathic intracranial hypertension. Curr Treat Options Neurol 2018; 20(7): 24
52. Francis, CE, Quiros, PA. Headache management in idiopathic intracranial hypertension. Int Ophthalmol Clin 2014; 54: 103-14.
53. Mollan, SP, Ali, F, Hassan-Smith, G, et al. Evolving evidence in adult idiopathic intracranial hypertension: pathophysiology and management. J Neurol Neurosurg Psychiatry 2016; 87: 982-92.
54. Friedman, DI, Liu, GT, Digre, KB. Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children. Neurology 2013; 81(13): 1159-65.
55. Wakerley, BR, Tan, MH, Ting, EY. Idiopathic intracranial hypertension. Cephalalgia 2015; 35(3): 248-61.
56. Orefice, G, Celentano, L, Scaglione, M, et al. Radioisotopic cisternography in benign intracranial hypertension of young obese women – a seven-case study and pathogenetic suggestions. Acta Neurol (Napoli) 1992; 14: 39-50.
57. Gomez, DG, Ehrmann, JE, Gordon Potts, D, et al. The arachnoid granulations of the newborn human: an ultrastructural study. Int J Dev Neurosci 1983; 1(2): 139-47.
58. Osaka, K, Handa, H, Matsumoto, S, et al. Development of the cerebrospinal fluid pathway in the normal and abnormal human embryos. Childs Brain 1980; 6(1): 26-38.
59. Johnston, M, Zakharov, A, Papaiconomou, C, et al. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cerebrospinal Fluid Res 2004; 1(1): 2.
60. Masakazu, M, Hajime, A. Evaluation of the production and absorption of cerebrospinal fluid. Neurol Med Chir (Tokyo) 2015; 55(8): 647-56.
61. Oreskovic, D, Rados, M, Klarica, M. Role of choroid plexus in cerebrospinal fluid hydrodynamics. Neuroscience 2017; 354: 69-87.
62. Riggeal, BD, Bruce, BB, Saindane, AM, et al. Clinical course of idiopathic intracranial hypertension with transverse sinus stenosis. Neurology 2013; 80: 289-95.
63. Dinkin, MJ, Patsalides, A. Venous sinus stenting for idiopathic intracranial hypertension: where are we now? Neurol Clin 2017; 35: 59-81.
64. Cserr, HF. Role of secretion and bulk flow of brain interstitial fluid in brain volume regulation. Ann N Y Acad Sci 1988; 529: 9-20.
65. Oreskovic, D, Klarica, M. The formation of cerebrospinal fluid: nearly a hundred years of interpretations and misinterpretations. Brain Res Rev 2010; 64(2): 241-62.
66. Bulat, M, Klarica, M. Recent insights into a new hydrodynamics of the cerebrospinal fluid. Brain Res Rev 2011; 65(2): 99-112.
67. Marakovic, J, Oreskovic, D, Rados, M, et al. Effect of osmolarity on CSF volume during ventriculo-aqueductal and ventriculo-cisternal perfusions in cats. Neurosci Lett 2010; 484(2): 93-7.
68. Daniels, AB, Liu, GT, Volpe, NJ, et al. Profiles of obesity, weight gain, and quality of life in idiopathic intracranial hypertension (pseudotumor cerebri). Am J Ophthalmol 2007; 143: 635-41.
69. Ko, MW, Chang, SC, Ridha, MA, et al. Weight gain and recurrence in idiopathic intracranial hypertension: a case-control study. Neurology 2011; 76: 1564-67.
70. Johnson, LB, Krohel, GM, Madsen, RW, et al. The role of weight loss and acetazolamide in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri). Ophthalmology 1998; 105: 2313-17.
71. Markey, KA, Mollan, SP, Jensen, RH, et al. Understanding idiopathic intracranial hypertension: mechanisms, management, and future decisions. Lancet Neurol 2016; 15(1): 78-91.
72. Radhakrishnan, K, Ahlskog, JE, Cross, SA, et al. Idiopathic intracranial hypertension (pseudotumor cerebri) – descriptive epidemiology in Rochester, Minn, 1976 to 1990. Arch Neurol 1993; 50(1): 78-80.
73. Raoof, N, Sharrack, B, Pepper, IM, et al. The incidence and prevalence of idiopathic intracranial hypertension in Sheffield, UK. Eur J Neurol 2011; 18(10): 1266-68.
74. Hamdallah, IN, Shamseddeen, HN, Getty, JL, et al. Greater than expected prevalence of pseudotumor cerebri: a prospective study. Surg Obes Relat Dis 2013; 9(1): 77-82.
75. Yri, HM, Jensen, RH. Idiopathic intracranial hypertension: clinical nosography and field-testing of the ICHD diagnostic criteria – a case-control study. Cephalalgia 2015; 35(7): 553-62.
76. Corbett, JJ, Jacobson, DM, Mauer, RC, et al. Enlargement of the blind spot caused by papilledema. Am J Ophthalmol 1988; 105(3): 261-65.
77. Grehn, F, Knorr-Held, S, Kommerell, G. Glaucomatouslike visual field defects in chronic papilledema. Albrecht Van Graefes Arch Klin Exp Ophthalmol 1981; 217(2): 99-109.
78. Friedman, DI. Papilledema and idiopathic intracranial hypertension. Continuum (Minneap Minn) 2014; 20: 857-76.
79. Wall, M, George, D. Visual loss in pseudotumor cerebri. Incidence and defects related to visual field strategy. Arch Neurol 1987; 44(2): 170-75.
80. Pastora-Salvador, N, Peralta-Calvo, J. Foster Kennedy syndrome: papilledema in one eye with optic atrophy in the other eye. CMAJ 2011; 183: 2135.
81. Micieli, JA, Al-Obthani, M, Sundaram, ANE. Pseudo-Foster Kennedy syndrome due to idiopathic intracranial hypertension. Can J Ophthalmol 2014; 49(4): e99-e102
82. Parafita-Fernandez, A, Sampil, M, Cores, C, et al. Foster Kennedy syndrome: an atypical presentation. Optom Vis Sci 2015; 92(12): e425-e430.
83. Kwancharoen, R, Blitz, AM, Tavares, F, et al. Clinical features of sellar and suprasellar meningiomas. Pituitary 2014; 17: 342-48.
84. Patil, A, Takkar, A, Goyal, M, et al. Sequential NAION presenting as pseudo Foster Kennedy syndrome. J Neurol Sci 2017; 376: 49-51.
85. Bansal, S, Dabbs, T, Long, V. Pseudo-Foster Kennedy syndrome due to unilateral optic nerve hypoplasia: a case report. J Med Case Rep 2008; 18: 2-86
86. Lam, BL, Morais, CG Jr., Pasol, J. Drusen of the optic disc. Curr Neurol Neurosci Rep 2008; 8: 404-08.
87. Tugcu, B, Ozdemir, H. Imaging methods in the diagnosis of optic disc drusen. Turk J Ophthalmol 2016; 46: 232-36.
88. Hamann, S, Malmqvist, L, Costello, F. Optic disc drusen: understanding an old problem from a new perspective. Acta Ophthalmol 2018 Apr 16; 1-12.
89. Sacks, JG, O’Grady, RB, Choromokos, E, et al. The pathogenesis of optic nerve drusen. A hypothesis. Arch Ophthalmol 1977; 95(3): 425-28.
90. Floyd, MS, Katz, BJ, Digre, KB. Measurement of the scleral canal using optical coherence tomography in patients with optic nerve drusen. Am J Ophthalmol 2005; 139(4): 664-69.
91. Atkins, EJ, Bruce, BB, Newman, NJ, et al. Treatment of nonarteritic anterior ischemic optic neuropathy. Surv Ophthalmol 2010; 55(1): 47-63.
92. Atkins, EJ. Nonarteritic anterior optic neuropathy. Curr Treat Options Neurol 2011; 13(1): 92-100.
93. Newman, NJ. The ischemic optic neuropathy decompression trial. Arch Ophthalmol 2007; 125(11): 1568-70.
94. Arnold, AC. Pathogenesis of nonarteritic anterior ischemic optic neuropathy. J Neurophthalmol 2003; 23(2): 157-63.
95. Deramo, VA, Sergott, RG. Augsberger, JJ, et al. Optic neuropathy as the first manifestation of elevated cholesterol in young patients. Ophthalmol 2003; 110(5): 1041-46.
96. Purvin, V, King, R, Kawasaki, A, et al. Anterior ischemic optic neuropathy in eyes with optic disc drusen. Arch Ophthalmol 2004; 122: 48-53.
97. Araz-Ersan, HB, Sayin, N, Bayramoglu, SE, et al. The effect of acute hypovolemia on the eye. Curr Eye Res 2018; 43(7): 949-54.
98. Tarantini, A, Faraoni, A, Menchini, F, et al. Bilateral simultaneous nonarteritic anterior ischemic optic neuropathy after ingestion of sildenafil for erectile dysfunction. Case Rep Med 2012; 2012: 747658.
99. Suarez-Fernandez, MJ, Clariana-Martin, A, Mencia-Gutierrez, E, et al. Bilateral anterior optic neuropathy after bilateral neck dissection. Clin Ophthalmol 2010; 4: 95-100.
100. Niro, A, Sborgia, G, Sbrogia, A, et al. Hyperhomocysteinemia in bilateral anterior ischemic optic neuropathy after conventional coronary artery bypass graft: a case report. J Med Case Rep 2018; 12: 11.
101. Nakao, K, Mizushima, Y, Abematsu, N, et al. Anterior ischemic optic neuropathy associated with Vogt-Koyanagi-Harada disease. Graefes Arch Clin Exp Ophthalmol 2009; 247(10): 1417-25.
102. Moorthy, RS, Inomata, H, Rao, NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol 1995; 39(4): 265-92.
103. Ohno, S, Minakawa, R, Matsuda, H. Clinical studies of Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol 1998; 32(3): 334-43.
104.Yokoyama, A, Ohta, K, Kojima, H. Vogt-Koyanagi-Harada disease masquerading as anterior ischaemic optic neuropathy. Br J Ophthalmol 1999; 83(1): 123
105.Abematsu, N, Shimonagano, Y, Nakao, K, et al. A case of anterio ischemic optic neuropathy associated with Vogt-Koyanagi-Harada disease. Nippon Ganka 2006; 110(8): 601-06.
106. Du, L, Kijlstra, A, Yang, P. Vogt-Koyanagi-Harada disease: novel insights into pathophysiology, diagnosis and treatment. Prog Retin Eye Res 2016; 52: 84-111.
107. Nguyen, HS, Haider, KM, Ackerman, LL. Unusual cases of papilledema: two illustrative cases. Surg Neurol Int 2013; 4: 60.
108. Waddell, LA, Greig, J, Mascarenhas, M, et al. The accuracy of diagnostic tests for Lyme disease in humans, a systematic review and meta-analysis of north American research. PLoS One 2016; 11(12): 1-23.
109. Fearon, M. The laboratory diagnosis of HIV infections. Can J Infect Dis Med Microbiol 2005; 16(1): 26-30.
110. Verma, R, Sarkar, S, Garg, RK, et al. Ophthalmological manifestation in patients of tuberculous meningitis. QJM 2019; 112(6): 409-419
111. Kelly, AM. Tuberculosis. Nurs Clin North Am 2019; 54(2): 193-205.
112. LoBue, PA, Mermin, JH. Latent tuberculosis infection: the final frontier of tuberculosis elimination in the USA. Lancet Infect Dis 2017; 17(10): e327-e333.
113. Browning, DJ. Posterior segment manifestations of active ocular syphilis, their response to a neurosyphilis regimen of penicillin therapy, and the influence of human immunodeficiency virus status on the response. Ophthalmology 2000; 107(11): 2015-23.
114. Goza, M, Kulwicki, B, Akers, JM, et al. Syphilis screening: a review of the syphilis health check rapid immunochromatographic test. J Pharm Technol 2017; 33(2): 53-9.
115. Valour, F, Senechal, A, Dupieux, C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist 2014; 7: 183-97.
116. Akhaddar, A, Elouennass, M, Baallal, H, et al. Focal intracranial infections due to Actinomyces species in immunocompetent patients: diagnostic and therapeutic challenges. World Neurosurg 2010; 74(2-3): 346-50.
117. Chiu, M, Moore, S. Bilateral optic disc swelling in Whipple’s disease. Clin Exp Ophthalmol 2017; 45(6): 641-43.
118. Bures, J, Kopacova, M, Douda, T, et al. Whipple’s disease: our own experience and review of the literature. Gastroenterol Res Pract 2013; 2013: 478349
119. Hafeez, MU, Ali, MH, Najib, N, et al. Ophthalmic manifestations of acute leukemia. Cureus 2019; 11(1): e3837.
120. Arber, DA, Orazi, A, Hasserjian, R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127(20): 2391-405.
121. Soman, S, Kasturi, N, Srinivasan, R, et al. Ocular manifestations in leukemias and their correlation with hematologic parameters at a tertiary care setting in south India. Ophthalmol Retina 2018; 2(1): 17-23.
122. Shen, K, Smith, SV, Lee, AG. Acute myelogenous leukemia presenting with uveitis, optic disc edema, and granuloma annulare: case report. Can J Ophthalmol 2016; 51(5): e153-e155.
123. Park, C, Suh, D, Ramey, A, et al. Isolated central nervous system primary acute monoblastic leukemia presenting as papilledema. Pediatr Blood Cancer 2016; 63(12): 2256-57.
124. Khadka, D, Sharma, AK, Shrestha, JK, et al. Ocular manifestations of childhood acute leukemia in a tertiary level eye centre of Kathmandu, Nepal. Nepal J Ophthalmol 2014; 6(2): 197-204.
125. Fernandez-Garcia, MA, Cantarin-Extremera, V, Andio-Catalan, M, et al. Secondary intracranial hypertension in pediatric patients with leukemia. Pediatri Neurol 2017; 77: 48-53.
126. Husebye, ES, Allolio, B, Arlt, W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med 2014: 275(2): 104-15.
127. Burton, C, Cottrell, E, Edwards, J. Addison’s disease: identification and management in primary care. Br J Gen Pract 2015; 62(638): 488-90.
128. Romano, N, Federici, M, Castaldi, A. Imaging of cranial nerves: a pictorial overview. Insights Imaging 2019; 10(1): 33
129. Feola, AJ, Coudrillier, B, Mulvihill, J, et al. Deformation of the lamina cribrosa and optic nerve due to changes in cerebrospinal fluid pressure. Invest Ophthalmol Vis Sci 2017; 58(4): 2070-78.
130. Sheth, S, Branstetter, BF IV, Escott, EJ. Appearance of normal cranial nerves on steady-state free precession MR images 2009; 29(4): 1045-55.
131. Yousry, I, Camelio, S, Wiesmann, M, et al. Detailed magnetic resonance imaging anatomy of the cisternal segment of the abducent nerve: Dorello’s canal and neurovascular relationships and landmarks. J Neurosurg 1999; 91(2): 276-83.
132. Goodwin, D. Differential diagnosis and management of acquired sixth cranial nerve palsy. Optometry 2006; 77(11): 534-39.
133. Reid, JE, Reem, RE, Aylward, SC, et al. Sixth nerve palsy in paediatric intracranial hypertension. Neuroophthalmology 2016; 40(1): 23-27.
134. Wall, M, George, D. Idiopathic intracranial hypertension – a prospective study of 50 patients. Brain 1991; 114(Pt 1A): 155-80.
135. Kapoor, A, Beniwal, V, Beniwal, S, et al. Isolated clival metastasis as the cause of abducens nerve palsy in a patient of breast carcinoma: a rare case report. Indian J Ophthalmol 2015; 63(4): 354-57.
136. Calandrelli, R, D’Apoloito, G, Panfili, M, et al. Costello syndrome: analysis of the posterior cranial fossa in children with posterior fossa crowding. Neuroradiol J 2015; 28(3): 254-58.
137. McCafferty, B, McClelland, CM, Lee, MS. The diagnostic challenge of evaluating papilledema in the pediatric patient. Taiwan J Ophthalmol 2017; 7(1): 15-21.
138. Padhye, LV, Van Stavern, GP, Sharma, A, et al. Association between visual parameters and neuroimaging features of idiopathic intracranial hypertension. J Neurol Sci 2013;332(1-2): 80-5.
139. Kupersmith, MJ, Sibony, P, Mandel, G, et al. Optical coherence tomography of the swollen optic nerve head: deformation of the peripapillary retinal pigment epithelium layer in papilledema. Invest Ophthalmol Vis Sci 2011; 52(9): 6558-64.
140. Burkett, JG, Ailanai, J. An up to date review of pseudotumor cerebri syndrome. Curr Neurol Neurosci Rep 2018; 18(6): 33.
141. Agid, R, Shelef, I, Scott, JN, et al. Imaging of the intracranial venous system. Neurologist 2008; 14(1): 12-22.
142. Roland, T, Jacobs, J, Rappaport, A, et al. Unenhanced brain CT is useful to decide on further imaging in suspected venous sinus thrombosis. Clin Radiol 2010; 65(1): 34-39.
143. Korein, J, Cravioto, H, Leicach, M. Reevaluation of lumbar puncture; a study of 129 patients with papilledema or intracranial hypertension. Neurology 1959; 9(4): 290-97.
144. Duffy, GP. Lumbar puncture in spontaneous subarachnoid haemorrhage. Br Med J (Clin Res Ed) 1982; 285(6349): 1163-64.
145. Duffy, GP. Lumbar puncture in the presence of raised intracranial pressure. Br Med J 1969; 1(5641): 407-09.
146. Mollan, SP, Davies, B, Silver, NC, et al. Idiopathic intracranial hypertension: consensus guidelines on management. J Neurol Neurosurg Psychiatry 2018; 89(10): 1088-1100.
147. Gertsl, L, Schoppe, N, Albers, L, et al. Pediatric idiopathic intracranial hypertension – is the fixed threshold value of elevated LP opening pressure set too high? Eur J Paediatr Neurol 2017; 21(6): 833-41.
148. Rangwala, LM, Liu, GT. Pediatric idiopathic intracranial hypertension. Surv Ophthalmol 2007; 52(6): 597-617.
149. Eidlitz-Markus, T, Stiebel-Kalish, H, Rubin, Y, et al. CSF pressure measurement during anesthesia: an unreliable technique. Paediatr Anaesth 2005; 12: 1078-82.
150. Cartwright, C, Igbaseimokumo, U. Lumbar puncture opening pressure is not a reliable measure of intracranial pressure in children. J Child Neurol 2015; 2: 170-73.
151. Ozturk, Z, Atalay, T, Arhan, E, et al. The efficacy of orbital ultrasonography and magnetic resonance imaging findings with direct measurement of intracranial pressure in distinguishing papilledema from pseudopapilledema. Childs Nerv Syst 2017; 33: 1501-07.
152. Braun, A, Doniger, SJ. Point-of-care ultrasonography for the identification of 2 children with optic disc drusen mimicking papilledema. Pediatr Emerg Care 2014; 30(7): 505-07.
153. Kurz-Levin MM, Landau, K. A comparison of imaging techniques for diagnosing drusen of the optic nerve head. Arch Ophthalmol 1999; 117: 1045-49.
154. Arbabi, EM, Feamley, TE, Carrim, ZI. Drusen and the misleading optic disc. Pract Neurol 2010; 10: 27-30.
155. Passi, N, Degnan, AJ, Levy, LM. MR imaging of papilledema and visual pathways: effects of increased intracranial pressure and pathophysiologic mechanisms. AJNR Am J Neuroradiol 2013; 5: 919-24.
156. Seitz, H, Held, P, Strozer, M, et al. Magnetic resonance imaging in patients diagnosed with papilledema: a comparison of 6 different high-resolution T1- and T2-weighted 3-dimensional and 2-dimensional sequences. J Neuroimaging 2002; 2: 164-71.
157. Watanabe, A, Kinouchi, H, Horikoshi, T, et al. Effect of intracranial pressure on the diameter of the optic nerve sheath. J Neurosurg 2008; 2: 255-58.
158. Maralani, PJ, Hassanlou, M, Torres, C, et al. Accuracy of brain imaging in the diagnosis of idiopathic intracranial hypertension. Clin Radiol 2012; 7: 656-63.
159. Shofty, B, Ben-Sira, L, Constantini, S, et al. Optic nerve sheath diameter on MR imaging: establishment of norms and comparison of pediatric patients with idiopathic intracranial hypertension with healthy controls. AJNR Am J Neurradiol 2012; 2: 366-69.
160. Lim, MJ, Pushparajah, K, Jan, W, et al. Magnetic resonance imaging changes in idiopathic intracranial hypertension in children. J Child Neurol 2010; 3: 294-99.
161. Hoffmann, H, Huppertz, HJ, Schmidt, C, et al. Morphometric and volumetric MRI changes in idiopathic intracranial hypertension. Cephalalgia 2013; 13: 1075-84.
162. Nicholson, P, Brinjikji, W, Radovanovic, I, et al. Venous sinus stenting for idiopathic intracranial hypertension: a systematic review and meta-analysis. J Neurointerv Surg 2019; 11(4): 380-85.
163. McCarthy, KD, Reed, DJ. The effect of acetazolamide and furosemide on cerebrospinal fluid production and choroid plexus carbonic anhydrase activity. J Pharmacol Exp Ther 1974; 189: 194-201.
164. Satti, SR, Leishangthem, L, Chaudry, MI. Meta-analysis of CSF diversion procedures and dural venous sinus stenting in the setting of medically refractory idiopathic intracranial hypertension. AJNR Am J Neuroradiol 2015; 36: 1899-1904.
165. Alsuhaibani, AH, Carter, KD, Nerad, JA, et al. Effect of optic nerve sheath fenestration on papilledema of the operated and the contralateral nonoperated eyes in idiopathic intracranial hypertension. Ophthalmology 2011; 118(2): 412-14.
166. Levin, AA, Hess, D, Hohler, AD. Treatment of idiopathic intracranial hypertension with gastric bypass surger. Int J Neurosci 2015; 125(1): 78-80.
167. Fridley, J, Foroozan, R, Sherman, V, et al. Bariatric surgery for the treatment of idiopathic intracranial hypertension. J Neurosurg 2011; 114: 34-39.
168. Newborg, B. Pseudotumor cerebri treated by rice reduction diet. Arch Intern Med 1974; 133: 802-07.
169. Kupersmith, MJ, Gamell, L, Turbin, R, et al. Effects of weight loss on the course of idiopathic intracranial hypertension in women. Neurology 1998; 50: 1094-98.
170. Wong, R, Madill, SA, Pandey, P, et al. Idiopathic intracranial hypertension: the association between weight loss and the requirement for systemic treatment. BMC Ophthalmol 2007; 7: 15.
171. Smith, SV, Friedman, DI. The idiopathic intracranial hypertension treatment trial: a review of the outcomes. Headache 2017; 557(8): 1303-10.
172. Weisberg, LA. Benign intracranial hypertension. Medicine (Baltimore) 1975; 54(3): 197-207.
173. Paterson, R, DePasquale, N, Mann, S. Pseudotumor cerebri. Medicine (Baltimore) 1961; 40: 85-99.
174. Weisberg, LA, Chutorian, AM. Pseudotumor cerebri of childhood. Am J Dis Child 1977; 131(11): 1243-48.

